Why is Quality Control Important in Genomics?
Posted on November 11, 2024
In this series, we are going to delve into a critical aspect of genomic epidemiology: the impact of sequencing and assembly errors on our interpretations. As you know, high-throughput sequencing has revolutionized our ability to track and understand infectious diseases. However, the accuracy of our epidemiological insights heavily relies on the quality of the sequencing and assembly processes. Errors in these processes can significantly distort our understanding of pathogen dynamics, leading to misleading conclusions about genetic diversity and transmission pathways. The type of issues include:
-
Inflating Genetic Diversity: Sequencing errors can introduce artificial nucleotide differences between sequences, leading to an overestimation of genetic diversity. A cluster of infections might appear more genetically diverse than it actually is, suggesting multiple sources of introduction rather than a single outbreak.
-
False Exclusion of Isolates and Incorrect Phylogenetic Placement: Errors can result in isolates being placed on incorrect branches of a phylogenetic tree. This can lead to the erroneous conclusion that certain isolates are not part of an outbreak when they actually are, complicating efforts to trace transmission pathways.
-
Misinterpretation of Transmission Dynamics: Erroneous sequences can disrupt the reconstruction of transmission chains, leading to inaccurate mappings of how a pathogen spreads through a population. Misidentifying a case as a new introduction rather than a continuation of an existing transmission chain.
The real effect of poor data
In this example, I selected 12 Salmonella enterica ser. Choleraesuis and created "a poorly assembled" genome from one of them (SAL_FC0090AA_AS). I then created a neighbour joining tree based on average nucleotide idenitity using mashtree to show you the effect. This is a common tool for creating a tree to show the similarity between genomes. BAD_FC0090AA_AS.result.fasta is a clear outlier, and no where near FC0090AA_AS.result.fasta which it was based on. If this analysis method was capable of handling the poorly assembled genome we should see the two genome together. This amount of difference between BAD_FC0090AA_AS.result.fasta and the others, is enough to change our intepretation. For instance, if the other genomes belonged to an outbreak, would we consider BAD_FC0090AA_AS.result.fasta part of that outbreak too?

This error won't be so pronounced in real data. And you won't be able to clearly spot it by eye in the final analysis. You need to be confident about the data quality from checking upstream.
A very special Salmonella Typhi
Someone once came to me with some results similar to the table below. The black means the genome had AMR determinants that would confer that resistance, white means absence. They were looking at multi-drug resistant Salmonella enterica serovar Typhi and found that one of their samples had a special profile of predicted AMR determinants that included extra mechanisms. They were very excited that they had found something new and wonderful and wanted me to just sanity check it.

I did a basic check of the taxonomic classification of the sample and it came back - Klebsiella pneumoniae. It was not a special Typhi, but a run of the mill Klebsiella that had been picked up by mistake.
The null result
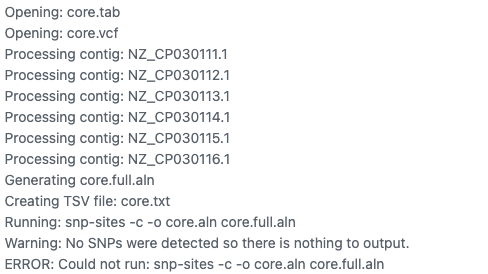
The most common manifestation of sequencing and genome assembly errors is that a downstream tool just doesn't work. Here is an error thrown by snippy when given poor quality data.
This content was prepared as part of the GenEpi-BioTrain programme funded by ECDC. The GenEpi-BioTrain programme is an interdisciplinary course in genomic and epidemiology, which was held at Institut Pasteur between May 27th and June 7th, 2024.
Genomic Quality Control
A comprehensive guide to quality control in bacterial genomics, from sequencing to assembly